
Ricardo A. Feldman, PhD
- Academic Title: Professor
- Primary Appointment: Microbiology and Immunology
- Email: rfeldman@som.umaryland.edu
- Location: HH, 320B
- Phone (Primary): 410-706-4197 (office)
- Phone (Secondary): 410-706-4198 (lab)
- Fax: 410-706-2129
Biosketch
Dr. Ricardo A. Feldman is a Professor at the UMSOM Department of Microbiology and Immunology.
Dr. Feldman received his PhD from the Department of Cell Biology at New York University School of Medicine, working with Drs. Takashi Morimoto and David Sabatini. After that he was a post-doctoral fellow at the Rockefeller University, in the laboratory of Dr. Hidesaburo Hanafusa. In Dr. Hanafusa's lab, he identified several new tyosine kinase oncogenes that had been captured by RNA tumor viruses, and identified their cellular homologs. Dr. Feldman then moved to NCI where he worked in the laboratory of Dr. Doug Lowy. He then joined the faculty of the UMSOM Department of Microbiology and Immunology.
Dr. Feldman is a also a founding member of The Center for Stem Cell Biology and Regenerative Medicine.
Research
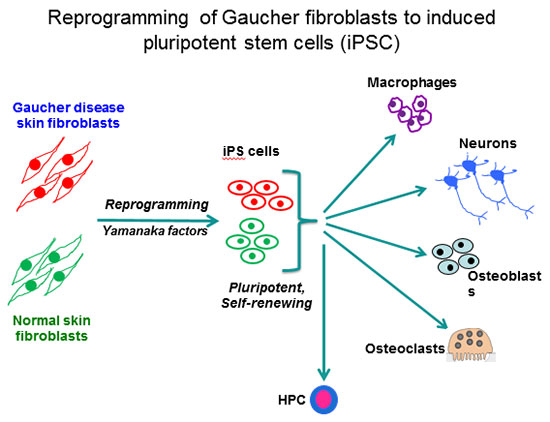
Our laboratory is using human induced pluripotent stem cells (iPSC) for disease modeling and drug discovery. By viral-mediated introduction of the 4 Yamanaka factors (Oct4, Sox2, Klf4 and c-Myc) we have reprogrammed patient fibroblasts into iPSC representative of all 3 clinical subtypes of Gaucher disease. This is an autosomal recessive disorder caused by mutations in the acid beta-glucocerebrosidase (GCase) gene (GBA1). GCase deficiency results in the accumulation of glucosylsphingolipids, leading to phenotypic abnormalities in a number of cell types, including macrophages, neuronal cells, osteoblasts, and hematopoietic cells.
The enzyme deficiency in Gaucher patients results in hepatosplenomegaly, hematologic abnormalities, bone disease, and there is a 50-fold increased propensity for development of B cell lymphomas and multiple myeloma. The more severe mutations in types 2 and 3 Gaucher disease also cause neurological manifestations that are fatal. The importance of this enzyme extends well beyond Gaucher disease. 7% of all Parkinson’s disease (PD) cases also have heterozygote mutations in GBA1, making them the highest known risk factor for PD. By directed differentiation of patient-derived iPSC to the affected cell types, we have recapitulated characteristic hallmarks of the disease, and obtained new mechanistic insights with important clinical implications for the treatment of GD and GBA1-associated PD.
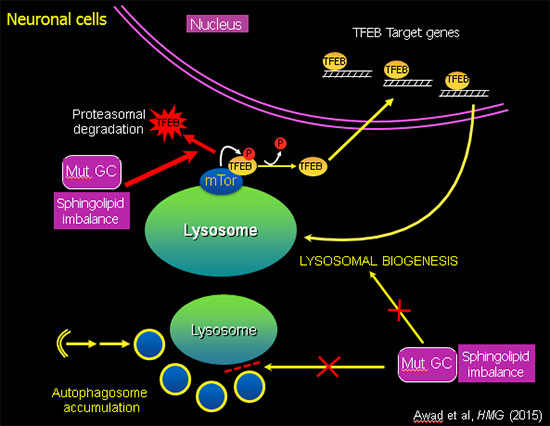
Sphingolipid imbalance caused by mutant GCase interferes with lysosomal biogenesis.
Using this unique "disease-in-a-dish" iPSC model, we found that the abnormal phenotype of Gaucher macrophages is not merely due to an accumulation of lipid, but there is also a defect in intracellular membrane transport, which blocks the flow of cargo (phagocytosed material, protein aggregates, organelles for recycling) to the lysosome. The mutant iPSC-derived macrophages exhibit a striking delay in clearing of phagocytosed red blood cells, which recapitulates a characteristic hallmark of the disease (Panicker et al., PNAS 2012). In mutant iPSC-derived neurons there is an autophagy block that prevents fusion of autophagosomes with lysosomes, leading to neuronal cell death. Analysis of the mechanisms involved showed that mutant GBA1 interferes with lysosomal biogenesis by downregulation and destabilization of TFEB, the master regulator of lysosomal and autophagy genes (Awad et al., Hum Mol Genet, 2015). Importantly, recombinant GCase, which is used in enzyme replacement therapy (ERT), and drugs used in substrate reduction therapy (SRT) reverse these phenotypes, showing the utility of this patient-derived iPSC system to evaluate the therapeutic efficacy of new agents in the treatment of sphingolipidoses.
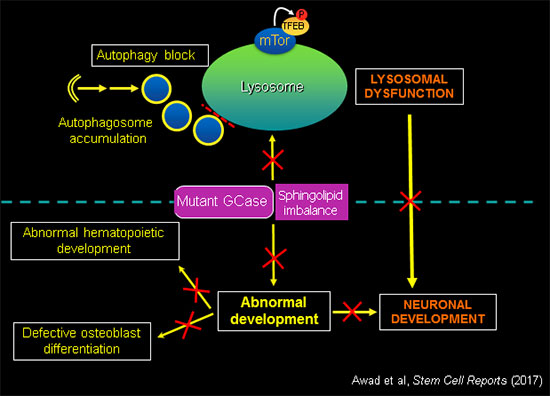
GCase activity is essential for normal development.
In addition to deregulation of essential lysosomal functions, mutant GBA1 causes developmental defects, recapitulating the anemias, bone disease and neurodegeneration observed in patients. Using the tools of molecular genetics we are elucidating the mechanisms by which GBA1 mutations and sphingolipid imbalance interfere with critical developmental pathways in different cell types affected by the disease.
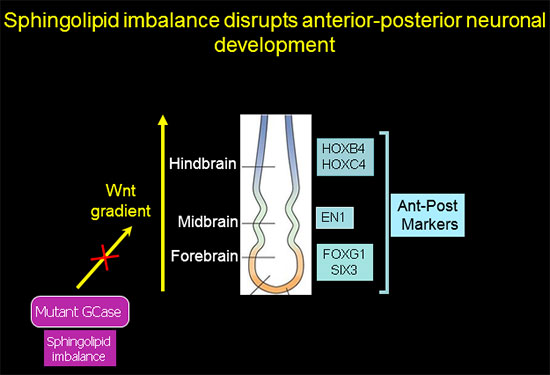
Sphingolipid imbalance interferes with anterior-posterior specification in developing brain due to downregulation of canonical Wnt/β-catenin signaling.
Anterior-posterior neuronal development is regulated by a Wnt gradient. Using iPSC from patients harboring severe GBA1 mutations we found that mutations in GCase that cause elevation in glucosylceramide and its cytotoxic metabolite glucosylsphingosine skew the normal patterning of early neuronal progenitors. This results in decreased survival of hindbrain, and midbrain dopaminergic neuronal progenitors, and overrepresentation of forebrain neuronal progenitors.
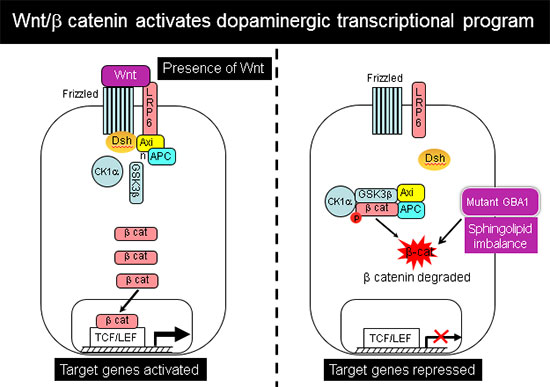
Further analysis showed that this abnormal developmental pattern was due to downregulation of canonical Wnt/β-catenin signaling, which is essential for hindbrain and midbrain development. Exogenous Wnt activation with small molecules rescued the mutant phenotype in 2-D cultures and in 3-D brain organoids, and restored differentiation to midbrain dopaminergic neurons (Awad et al., Stem Cell Reports 2017). Our work shows for the first time a Wnt-dependent molecular link between GD and GBA1-associated Parkinson's disease, providing a new therapeutic target that is amenable to pharmacological intervention.
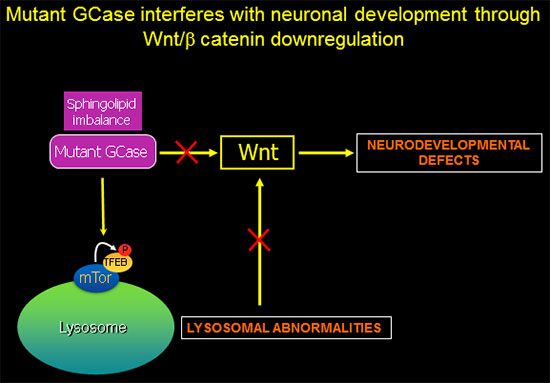
GBA1-associated lysosomal dysfunction is directly linked to Wnt downregulation and neurodevelopmental abnormalities.
In recently published work (Awad et al., Stem Cell Reports 2017), we showed that reduced sequestration of GSK3β by a defective endolysosomal compartment in the mutant cells leads to increased β-catenin degradation and attenuation of the canonical Wnt signal. Thus, the lysosomal abnormalities caused by GCase deficiency and sphingolipid imbalance are directly involved in downregulation of essential neurodevelopmental pathways. We additionally found that there are multiple nodes of cross-talk between lysosomal function and Wnt signaling.
Mutant GBA1 iPSC-derived osteoblasts have developmental and lysosomal defects that impair bone matrix deposition.
The Wnt-dependent developmental defects caused by GCase deficiency are not limited to neurons. Differentiation of mutant iPSC to osteoblasts showed that these cells have developmental defects and lysosomal abnormalities that interfere with bone matrix deposition, reflecting the bone pathology seen in GD patients. GCase deficiency causes reduced expression of osteoblast differentiation markers, and bone matrix protein and mineral deposition are defective. Concomitantly, canonical Wnt/β-catenin signaling in the mutant osteoblasts was downregulated, whereas pharmacological Wnt activation rescued GD osteoblast differentiation and bone matrix deposition. Importantly, incubation with recombinant GCase (rGCase) rescued the differentiation and bone-forming ability of GD osteoblasts, demonstrating that the abnormal GD phenotype was indeed caused by GCase deficiency (Panicker et al., Hum Mol Genet 2018). GD osteoblasts were also defective in their ability to carry out Ca2+-dependent exocytosis, a lysosomal function that is necessary for bone matrix deposition. Thus, GCase enzymatic activity is also required for the differentiation and bone-forming activity of osteoblasts. Furthermore, the rescue of bone matrix deposition by pharmacological activation of Wnt/β-catenin in GD osteoblasts uncovers a new therapeutic target for the treatment of bone abnormalities in GD including osteoporosis.
GCase deficiency causes aberrant hematopoiesis.
Normal GCase activity is also required for hematopoietic development (Sgambato et al., Stem Cells Translational Medicine 2015). Mutations in this enzyme result in decreased erythroid potential and aberrant myelopoiesis, recapitulating the anemias seen in GD patients, again demonstrating the utility of patient-derived iPS for disease modeling and therapeutic development.
Identification of a novel lysosome-based sphingolipid-sensing mechanism using patient-derived iPSC.
Our laboratory is currently identifying the molecular components of a lysosome-based machinery that is responsible for the deleterious effects of sphingolipid imbalance in patient-derived iPSC harboring mutations in GBA1. The new targets identified are amenable to pharmacological intervention.
Previous work in the laboratory involved the use transgenic mice (SCL-TVA), which allowed targeted delivery of genes to hematopoietic and vascular endothelial stem cells in the intact animal. One application of this gene delivery system is in vivo labeling of long-term self-renewing stem cells with markers such as luciferase. This allowed us to visualize the location of hemangioblasts and hematopoietic stem cells in their natural locations by bioluminescence imaging of live animals, and to follow the fate and mobilization of these stem cells in response to injury or biotherapeutic agents, in real time. This flexible experimental system circumvents the problems associated with conventional in vitro manipulation of bone marrow stem cells and transplantation into lethally irradiated animals.
A goal of our laboratory is to combine our expertise in reprogramming technology and targeted delivery of genes to specific tissues, for in vivo reprogramming, gene therapy, and regenerative medicine.
Research/Clinical Keywords
Human induced pluripotent stem cells (iPSC) for disease modeling, drug discovery and regenerative medicine; lysosomal storage diseases; Gaucher disease; Parkinson's disease; neuronal development; dopaminergic development; neurodegeneration; Wnt pathway; GSK3β; β-catenin; glucocerebrosidase; sphingolipids; mTOR; TFEB; enzyme replacement therapy (ERT); substrate reduction therapy (SRT), mesenchymal stem cells (MSC); osteoblasts; bone disease; macrophages; hematopoietic development.
Publications
![]()
Complete list of Publications:
https://www.ncbi.nlm.nih.gov/myncbi/1bE58egne9vkN/bibliography/public/
Panicker LM, Gomez, T, Srikanth, M, Miller, D, Andrews, NW, and Feldman, RA. Gaucher disease iPSC-derived osteoblasts have developmental and lysosomal defects that impair bone matrix deposition. (2018). Hum Mol Genet, 27: 811–822. PMID: 29301038.
Awad O, Panicker LM, Deranieh RM, Srikanth MP, Brown R, Voit A, Peesay T, Park TS, Zambidis ET, and Feldman RA. (2017). Altered differentiation potential of Gaucher disease iPSC-neuronal progenitors due to Wnt/β-catenin downregulation. Stem Cell Reports, (2017) Dec 12;9(6):1853-1867. doi: 10.1016/j.stemcr.2017.10.029. Epub 2017 Nov 30. PubMed PMID:29198828.
Jeon, OH, Panicker, LM, Lu, Q, Chae, JJ, Feldman, RA. and Elisseeff, JH. Human iPSC-derived osteoblasts and osteoclasts together promote bone regeneration in 3D biomaterials. (2016). Sci Rep May 26;6:26761. doi: 10.1038/srep26761. PMC4881234.
Gramlich PA, Westbroek W, Feldman RA, Awad O, Mello N, Remington MP, Sun Y, Zhang W, Sidransky E, Betenbaugh MJ, Fishman PS. A Peptide-Linked Recombinant Glucocerebrosidase for Targeted Neuronal Delivery: Design, Production, and Assessment. (2016). J Biotechnol. 221: 1-12. doi: 10.1016/j.jbiotec.2016.01.015. PubMed PMID: 26795355.
Awad, O., Sarkar S., Panicker L. M., Miller D., Zeng, X., Sgambato, J.A, Lipinski, M. M., and Feldman, R.A. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSCs-derived neuronal cells. Hum Mol Genet. 2015 Jul 28. pii: ddv297. [Epub ahead of print]. Supplement
Spiller KL, Wrona EA, Romero-Torres S, Pallotta I, Graney PL, Witherel CE, Panicker LM, Feldman RA, Urbanska AM, Santambrogio L, Vunjak-Novakovic G, Freytes DO. Differential Gene Expression in Human, Murine, and Cell Line-derived Macrophages upon Polarization. (2015). Exp Cell Res. pii: S0014-4827(15)30121-X. doi: 10.1016/j.yexcr.2015.10.017. [Epub ahead of print] PubMed PMID: 26500109.
Sgambato, J.A, Park T. S., Miller D., Panicker, L. M., Sidransky, E., Lun, Y., Awad, O., Bentzen, S. M., Zambidis E.T., and Feldman R.A. (2015). Gaucher disease induced pluripotent stem cells display decreased erythroid potential and aberrant myelopoiesis. Stem Cells Transl Med. 4: 878-86. PDF
Panicker L. M., Miller D., Awad O., Bose, V., Lun, Y., Park T. S., Zambidis E. T., Sgambato J. A., and Feldman R. A. (2014). Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells. 32: 2338-2349. PDF
Park T. S., Bhutto I., Zimmerlin L., Huo, J. S., Nagaria P., Miller D., Jalil R.A., Talbot C., Aguilar J., Merges C., Reijo-Pera R., Feldman RA. Rassool F., Cooke J., Lutty G., Zambidis E.T. (2014). Vascular Progenitors from Cord Blood-Derived iPSC Possess Augmented Capacity for Regenerating Ischemic Retinal Vasculature. Circulation 129: 359-72.
Panicker L.M., Miller D., Park T.S., Patel B., Azevedo J.L., Awad O., Masood, M.A., Veenstra, T.D., Goldin E., Stubblefield, B., Tayebi, N., Polumuri S.K., Vogel S.N., Sidransky, E., Zambidis E.T., and Feldman R.A. (2012). Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease.Proc Natl Acad Sci USA. 109: 18054-9.
Azevedo, J.L., and Feldman, R.A. Tinkering with transcription factors uncovers plasticity of somatic cells. (2010). Genes & Cancer 1: 1089-99.
Xie, Y., Yin, T., Wiegraebe, W., He, X.C., Miller, D., Stark, D., Perko, K., Alexander, R., Schwartz, J., Grindley, J., Park, J., Haug, J., Wunderlich, J., Li, H., Zhang, S., Johnson, T., Feldman, RA, and Li, L. (2009) Detection of functional hematopoietic stem cell niche using real-time imaging. Nature, 457: 97-101.
- Comment in Nature Reports Stem Cells: Published online 15 January (2009).
- Comment in Cell, 136: 7 (2009).
Sausville J., Molinolo, A., Cheng, X., Frampton, J., Takebe N., Gutkind, J.S., and Feldman, RA. (2008). RCAS/SCL-TVA animal model allows targeted delivery of PyMT oncogene to vascular endothelial progenitorsin vivo, and results in hemangioma development. Clin. Cancer Res. 14: 3948-3955.
Kim, J., Ogata, Y., and Feldman, RA. (2003). Fes tyrosine kinase promotes survival and terminal granulocyte differentiation of factor-dependent myeloid progenitors (32D) and activates lineage-specific transcription factors. J. Biol. Chem. 278: 14978-14984.
Kim, J., and Feldman, RA. (2002). Activated Fes Protein Tyrosine Kinase Induces Terminal Macrophage Differentiation of Myeloid Progenitors (U937 cells) and Activation of the Transcription factor PU.1. Mol. Cell. Biol. 22: 1903-1918.
Jücker, M., Südel, K., Horn, S., Sickel, M., Wegner, W., Fiedler, W., and Feldman, RA. (2002). Expression of a mutated form of the p85 alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkin's lymphoma-derived cell line (CO). Leukemia 16: 894-901.