Traumatic spinal cord injury (SCI) causes tissue loss and associated neurological dysfunction due to both mechanical damage and secondary biochemical and physiological responses. Mechanisms of secondary injury include a complex cellular response through the activation and/or suppression of a large number of transcriptional pathways in a heterogeneous cell population. These mechanisms include neuronal cell death, loss of oligodendrocytes, inflammation and reactive astrogliosis. Effective clinical strategies will likely include either combination of selective inhibitors of secondary injury factors or single drugs that modulate multiple injury components. Since 2004, we have investigated the role of cell cycle pathways in the pathobiology of neurotrauma. Substantial experimental evidence, much from our laboratory, indicates that in post-mitotic neurons cell cycle activation (CCA) is associated with neuronal cell death, whereas in microglia or astrocytes it leads to cell proliferation and activation. Thus, identification of common molecular pathways, including the role of cell cycle proteins involved in both neuronal death and reactive gliosis, may help to clarify the pathobiology of CNS injury and lead to the elucidation of novel therapeutic targets.
SCI causes long-term disability including loss of motor and sensory function, as well as chronic neuropathic pain in up to 80% of patients that responds poorly to conventional analgesic therapies. We have been examining the role of specific CCA pathways not only in secondary spinal cord tissue damage after SCI, but also as a critical mechanism for development of central neuropathic pain. A highly synergistic collaboration between the Faden and Dorsey laboratories has linked the trkB.T1 receptor to posttraumatic hyperalgesia after SCI through regulation of cell cycle pathways. Separate collaborative studies with Asaf Keller’s laboratory demonstrated that hyperalgesia after SCI is also related to increased excitation within the dorsal thalamic nucleus; these effects are also reduced by treatment with a cell cycle inhibitor.
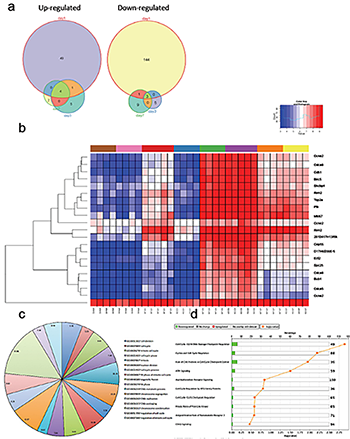 Fig 1.
Fig 1.
Cell cycle genes are upregulated in the spinal cord of trkB.T1+/+ mice early after SCI but not in the trkB.T1 null.
- Differential gene expression analysis over time demonstrates that the largest number of genes is altered early after SCI, at day one. The Venn diagrams show each time point, with the number of differentially expressed genes.
- Heat map of genes that cluster by genotype and time in trkB.T1-/- spinal cord compared with wildtype. The normalized expression of the summarized probe sets is shown. The histogram key indicates that blue genes are down regulated and red genes are up regulated.
- Gene ontology (GO) analysis. The pie chart lists the 17 significantly enriched GO terms in knockout versus wildtype spinal cord at day one. The numbers in each slice represent the degree of enrichment; all are statistically significant at FDR P < 0.02.
- Unbiased pathway analysis demonstrates significant down regulation of cell cycle pathways in trkB.T1-/- spinal cord compared with wildtype. All are significant per the –log10 p values shown as orange squares (n=4-7).
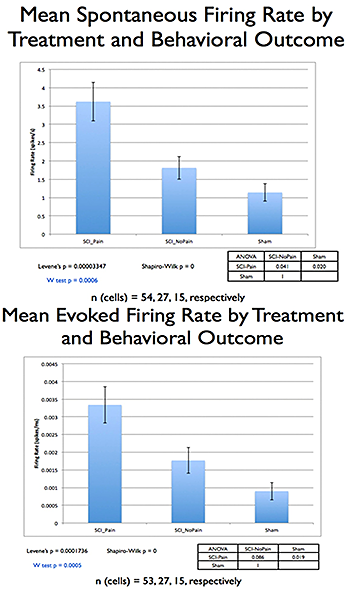
Fig 2.
Comparing SCI rats with or without post-injury hyperalgesia, and sham-operated controls, showed significant differences (p=0.005, Kruskal-Wallis ANOVA), with a significant correlation between increases in the posterior thalamic nucleus (PO) spontaneous/evoked activity and hyperalgesia.