High Throughput In Vivo Functional Validation of Candidate Congenital Heart Disease Genes in Drosophila
Zhu JY, Fu Y, Nettleton M, Richman A, and Han Z Elife 2017; 6. pii: e22617.
In the news:
Patient clinical features:
Patients came from a cohort of severe congenital heart disease (CHD) patients (Zaidi et al, 2013). The patient with a WDR5 genetic variant associated with CHD was described as showing connective tissue disease (CTD) pathology characterized by Tetralogy of Fallot (ToF) a complex heart defect including ventricular septal defect (VSD), pulmonary stenosis (PS), right ventricular hypertrophy and overriding aorta, with right aortic arch, aberrant left subclavian artery (LSA) and coronary abnormality. The patient had no extracardiac structural anomalies but did display abnormal neurodevelopment.
Patient genetic variant:
The authors (Zaidi et al, 2013) used exome sequencing of parent-offspring trios to identify de novo mutations associated with CHD. They found an excess of de novo mutation in genes involved in the production, removal or reading of histone 3 lysine 4 (H3K4) methylation, as well as in regulatory genes of H3K4 methylation (H3K4me). H3K4me is an activating mark found in promoters/enhancers of key developmental genes, important for directing selective gene activation in different cell lineages. Among the genetic variants they identified was a missense mutation in WDR5 (WDR5-K7Q variant), a component of the MLL2 H3K4 N-methyltransferase complex.
Footnote: Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, Kim R, Kline JK, Lee T, Leipzig J, Lopez A, Mane SM, Mitchell LE, Newburger JW, Parfenov M, Pe'er I, Porter G, Roberts AE, Sachidanandam R, Sanders SJ, Seiden HS, State MW, Subramanian S, Tikhonova IR, Wang W, Warburton D, White PS, Williams IA, Zhao H, Seidman JG, Brueckner M, Chung WK, Gelb BD, Goldmuntz E, Seidman CE, and Lifton RP. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013; 498(7453):220-223.
Precision Drosophila disease model:
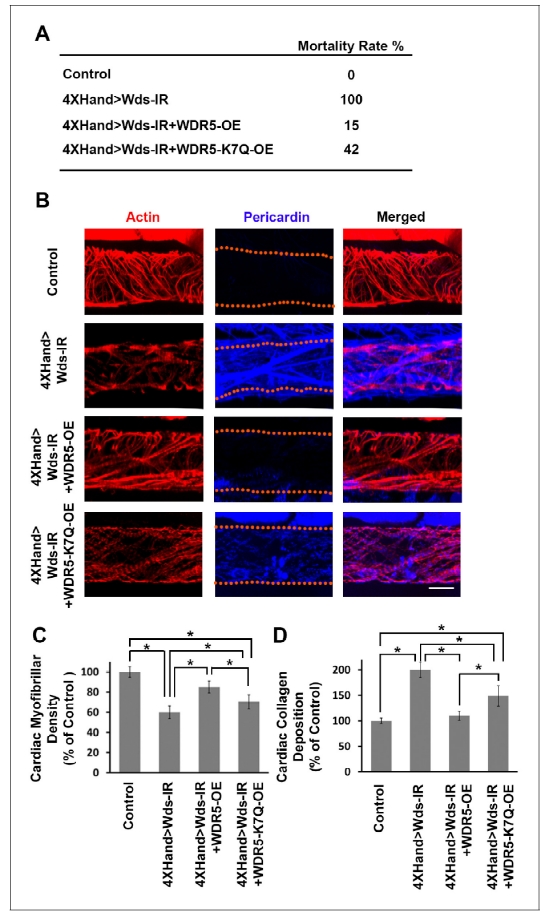
We developed a high-throughput in vivo model system to screen candidate genes identified in CHD patients. In essence, we apply RNAi-based gene silencing specific to Drosophila heart, then carry out quantitative assays for multiple cardiac phenotypes to identify genes essential for heart structure, function and development. We employed this platform combined with our ‘gene replacement’ strategy to demonstrate its use in evaluating cardiac phenotypes resulting from specific, patient-derived alleles of candidate disease genes. As proof-of-concept, we tested the ability of a reference and CHD-associated variant human WDR5 alleles to rescue heart phenotypes induced by silencing of the endogenous wds Drosophila homolog. Heart-specific silencing of wds caused 100% developmental lethality (Figure 1A) and abnormal heart morphology in late larvae characterized by reduced cardiac myofibers (Figure 1, B and C) and severe over-deposition of Pericardin (Figure 1, B and D). We found that simultaneously overexpressing reference human WDR5 allele reduced developmental lethality almost 7-fold, significantly restored cardiac myofibrillar density, and reduced Pericardin levels essentially to normal. By contrast, when the endogenous wds expression was ‘replaced’ by a CHD patient-derived WDR5-K7Q disease-variant allele, developmental lethality remained quite elevated, cardiac myofibrillar density remained abnormally low (relative both to control and reference WDR5 rescue), and Pericardin levels were lowered but still significantly higher than both control and reference WDR5 rescue. These observations illustrate the structure-function homologies between human and fly ‘heart genes’ that support use of Drosophila for functional validation of candidate CHD genes. Moreover, they demonstrate that the ‘gene replacement’ strategy can be used to quantitatively assess phenotypes induced by patient- specific alleles of candidate disease genes.